Write a super simple molecular dynamics program in python
Molecular dynamics calculation of 3 atoms
Let's write a molecular dynamics (MD) code that deals with only three atoms. Make it MD-like step by step.
1. Place atoms on the canvas
Write the initial arrangement and velocity of the three atoms in a file.
initial.d
15 10 0 0
20 10 0 0
20 15 0 0
Each line of initial. d consists of four items: the x-coordinate of the atom, the y-coordinate, the x-component of the velocity, and the y-component, and must be separated by a space.
An example of a program that opens the canvas so that an object can be drawn, reads the atom arrangement (and velocity) information from the file (initial.d), and arranges the atoms at the initial position is shown below. I also have a dummy button for later (here I do nothing when the button is pressed). Here, since initial.d has 3 rows, the number of atoms is 3, but the number of atoms can be increased by increasing the number of rows. In the program, the array size is increased each time each row is read. The unit of coordinates is Å, and the unit of velocity is Å / s.
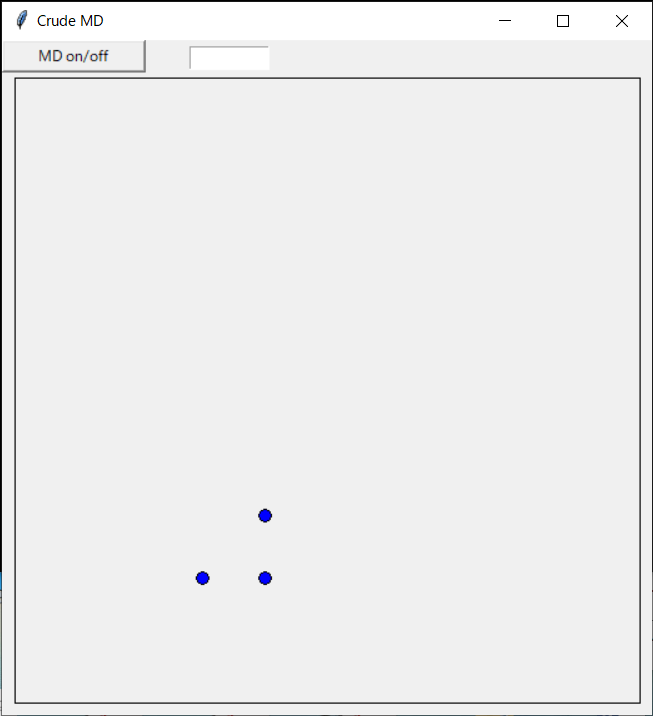
An example program is shown.
crudemd0.py
import tkinter as tk
def dummy(event):
print('button is pressed')
def drawatom(x,y):
scl=10
rad=5
x1=x0+x*scl
y1=y0+l-y*scl
canvas.create_oval(x1-rad,y1-rad,x1+rad,y1+rad, fill='blue')
# creation of main window and canvas
win = tk.Tk()
win.title("Crude MD")
win.geometry("520x540")
x0=10
y0=10
l=500
canvas = tk.Canvas(win,width=l+x0*2,height=l+y0*2)
canvas.place(x=0, y=20)
canvas.create_rectangle(x0,y0,l+x0,l+y0)
# declaration of arrays
rx = []
ry = []
vx = []
vy = []
fx = []
fy = []
# button sample (dummy)
button_dummy = tk.Button(win,text="dummy",width=15)
button_dummy.bind("<Button-1>",dummy)
button_dummy.place(x=0,y=0)
# read initial position and velocity
f = open('initial.d', 'r')
i = 0
for line in f:
xy = line.split(' ')
rx = rx + [float(xy[0])]
ry = ry + [float(xy[1])]
vx = vx + [float(xy[2])]
vy = vy + [float(xy[3])]
print(rx)
drawatom(rx[i],ry[i])
i = i + 1
win.mainloop()
2. Get the button event and move the atom
Added functions based on the above program. It detects the event that the button is pressed and moves the atom diagonally upward just continuously.
crudemd1.py
import tkinter as tk
def dummy(event):
global imd # to substitute value into variable
if (imd == 0 ):
imd = 1
else:
imd = 0
def drawatom(x,y):
x1=x0+x*scl
y1=y0+l-y*scl
return canvas.create_oval(x1-rad,y1-rad,x1+rad,y1+rad, fill='blue')
def moveatom(obj,x,y):
x1=x0+x*scl
y1=y0+l-y*scl
canvas.coords(obj,x1-rad,y1-rad,x1+rad,y1+rad)
# creation of main window and canvas
win = tk.Tk()
win.title("Crude MD")
win.geometry("520x540")
x0=10 # Origin
y0=10 # Origin
l=500 # Size of canvas
scl=10 # Scaling (magnification) factor
rad=5 # Radius of sphere
canvas = tk.Canvas(win,width=l+x0*2,height=l+y0*2)
canvas.place(x=0, y=20)
canvas.create_rectangle(x0,y0,l+x0,l+y0)
imd = 0 # MD on/off
# declaration of arrays
rx = []
ry = []
vx = []
vy = []
fx = []
fy = []
obj = []
# button sample (dummy)
button_dummy = tk.Button(win,text="dummy",width=15)
button_dummy.bind("<Button-1>",dummy)
button_dummy.place(x=0,y=0)
# Entry box (MD step)
entry_step = tk.Entry(width=10)
entry_step.place(x=150,y=5)
# read initial position and velocity
f = open('initial.d', 'r')
n = 0
for line in f:
xy = line.split(' ')
rx = rx + [float(xy[0])]
ry = ry + [float(xy[1])]
vx = vx + [float(xy[2])]
vy = vy + [float(xy[3])]
print(rx)
obj = obj + [drawatom(rx[n],ry[n])] # obj[] holds pointer to object
n = n + 1 # number of total atoms
print("number of atoms = ",n)
step = 0
stepend = 1000
while step < stepend:
if imd == 1:
for i in range(n):
rx[i] = rx[i] + 0.01
ry[i] = ry[i] + 0.01
moveatom(obj[i],rx[i],ry[i])
step = step + 1
entry_step.delete(0,tk.END)
entry_step.insert(tk.END,step)
win.update()
win.mainloop()
3. Turn MD on / off when the button is pressed
Further modified the above. The MD is switched on / off when the button is pressed. The interaction between atoms is calculated by Morse potential, and the atom is moved by the Verlet method.
crudemd2.py
import tkinter as tk
import math
def dummy(event):
global imd # to substitute value into variable
if (imd == 0 ):
imd = 1
else:
imd = 0
def drawatom(x,y):
x1=x0+x*scl
y1=y0+l-y*scl
return canvas.create_oval(x1-rad,y1-rad,x1+rad,y1+rad, fill='blue')
def moveatom(obj,x,y):
x1=x0+x*scl
y1=y0+l-y*scl
canvas.coords(obj,x1-rad,y1-rad,x1+rad,y1+rad)
def v(rang):
ep = 0.2703
al = 1.1646
ro = 3.253
ev = 1.6021892e-19
return ep*(math.exp(-2.0*al*(rang-ro))-2.0*math.exp(-al*(rang-ro))) *ev
def vp(rang):
ep = 0.2703
al = 1.1646
ro = 3.253
ev = 1.6021892e-19
return -2.0*al*ep*(math.exp(-2.0*al*(rang-ro))-math.exp(-al*(rang-ro))) *ev*1.0e10
# creation of main window and canvas
win = tk.Tk()
win.title("Crude MD")
win.geometry("520x540")
x0=10 # Origin
y0=10 # Origin
l=500 # Size of canvas
scl=10 # Scaling (magnification) factor
rad=5 # Radius of sphere
canvas = tk.Canvas(win,width=l+x0*2,height=l+y0*2)
canvas.place(x=0, y=20)
canvas.create_rectangle(x0,y0,l+x0,l+y0)
imd = 0 # MD on/off
# declaration of arrays
rx = [] # ang
ry = []
vx = [] # ang/s
vy = []
fx = [] # N
fy = []
epot = []
obj = []
dt = 1.0e-16 # s
wm = 1.67e-37 # 1e-10 kg
# button sample (dummy)
button_dummy = tk.Button(win,text="MD on/off",width=15)
button_dummy.bind("<Button-1>",dummy)
button_dummy.place(x=0,y=0)
# Entry box (MD step)
entry_step = tk.Entry(width=10)
entry_step.place(x=150,y=5)
# read initial position and velocity
f = open('initial.d', 'r')
n = 0
for line in f:
xy = line.split(' ')
rx = rx + [float(xy[0])]
ry = ry + [float(xy[1])]
vx = vx + [float(xy[2])]
vy = vy + [float(xy[3])]
fx = fx + [0]
fy = fy + [0]
epot = epot + [0]
obj = obj + [drawatom(rx[n],ry[n])] # obj[] holds pointer to object
n = n + 1 # number of total atoms
print("number of atoms = ",n)
step = 0
stepend = 100000
while step < stepend:
if imd == 1:
# Verlet(1)
for i in range(n):
rx[i] = rx[i] + dt * vx[i] + (dt*dt/2.0) * fx[i] / wm
ry[i] = ry[i] + dt * vy[i] + (dt*dt/2.0) * fy[i] / wm
vx[i] = vx[i] + dt/2.0 * fx[i] / wm
vy[i] = vy[i] + dt/2.0 * fy[i] / wm
# Force and energy
for i in range(n):
fx[i] = 0
fy[i] = 0
epot[i] = 0
for i in range(n):
for j in range(n):
if (i != j):
rr = math.sqrt((rx[i]-rx[j])**2 + (ry[i]-ry[j])**2)
drx = rx[i] - rx[j]
dry = ry[i] - ry[j]
fx[i] = fx[i]-vp(rr)/rr*drx
fy[i] = fy[i]-vp(rr)/rr*dry
epot[i] = epot[i]+v(rr)/2.0
# Verlet(2)
for i in range(n):
vx[i] = vx[i] + dt/2.0 * fx[i] / wm
vy[i] = vy[i] + dt/2.0 * fy[i] / wm
moveatom(obj[i],rx[i],ry[i])
step = step + 1
entry_step.delete(0,tk.END)
entry_step.insert(tk.END,step)
win.update()
win.mainloop()
Recommended Posts