2 herunter. Es gibt zwei Arten von Probensuchdaten für ChemTHEATER, Messdaten und Proben, aber dieses Mal werden wir beide verwenden.
Die ChemTHEATER-Bedienungsanleitung finden Sie auf der ChemTHEATER-Wiki-Website .
Die einfache Bedienungsanleitung lautet wie folgt.
- Klicken Sie in der Menüleiste auf der oberen Seite von ChemTEHATRE auf "Beispielsuche".
- Wählen Sie im Pulldown-Menü "Probentyp" die Option "Biotisch - Säugetiere - Meeressäuger".
- Wählen Sie den wissenschaftlichen Namen "Neophocaena phocaenoides" von Snameri aus dem Pulldown-Menü von "Scientific Name".
- Klicken Sie auf die Schaltfläche "Suchen".
- Klicken Sie auf die Schaltfläche "Proben exportieren (TSV)", um die Probenliste zu exportieren, und auf die Schaltfläche "Messdaten exportieren (TSV)", um die Messwertliste als tabulatorgetrennte Textdateien zu exportieren.
- Speichern Sie die exportierte Datei in einem beliebigen Verzeichnis und verwenden Sie sie für die nachfolgende Analyse.
data_file = "measureddata_20190826061813.tsv" #Ändern Sie die in die Variable eingegebene Zeichenfolge in den tsv-Dateinamen Ihrer Messdaten
chem = pd.read_csv(data_file, delimiter="\t")
chem = chem.drop(["ProjectID", "ScientificName", "RegisterDate", "UpdateDate"], axis=1) #Entfernen Sie doppelte Spalten, wenn Sie später mit Samples verbinden
sample_file = "samples_20190826061810.tsv" #Ändern Sie die in die Variable eingegebene Zeichenfolge in den tsv-Dateinamen Ihrer Beispiele
sample = pd.read_csv(sample_file, delimiter="\t")
Die heruntergeladene TSV-Datei wird von der Pandas-Funktion (read_csv) gelesen. Zu diesem Zeitpunkt muss der Pfad jeder Datei ("Datendatei" und "Beispieldatei" in der oberen Zelle) in einen für jede Umgebung geeigneten Dateinamen geändert werden. Der Beispielcode wird in einer Umgebung geschrieben, in der sich die Datendatei im selben Verzeichnis wie das Notizbuch befindet.
## Kapitel 3 Datenaufbereitung
Die diesmal verarbeiteten Daten sind eine Zusammenstellung der Messergebnisse verschiedener chemischer Substanzen. Weiterhin sind die Messergebnisse und die Probendaten gestreut. Mit anderen Worten, es ist schwierig zu handhaben, so wie es ist, daher ist es notwendig, es in eine Form zu formen, die leicht zu handhaben ist. Insbesondere sind die folgenden Operationen erforderlich.
Probe mit - chem.
- Extrahieren Sie diesmal nur die Messdaten der behandelten chemischen Substanzen.
- Löschen Sie Spalten, die für die Visualisierung nicht benötigt werden.
Abschnitt 3-1 Extraktion der erforderlichen Daten
Führen Sie zunächst die oben gezeigten Prozesse 1 und 2 aus.
Bindung ist die Arbeit des Hinzufügens eines Probendatenrahmens (Probe) einer Probe jeder chemischen Substanz zum Datenrahmen (chem) jeder chemischen Substanz. Hier wird der Prozess des Anhängens der Probendaten, die der Proben-ID jeder Chem-Daten entsprechen, an die rechte Seite der Chem durchgeführt.
df = pd.merge(chem, sample, on="SampleID")
In den diesmal verarbeiteten Daten wird jede chemische Substanz ziemlich detailliert klassifiziert. Sie können die Messergebnisse für jede chemische Substanz visualisieren. Dieses Mal möchten wir jedoch die Umrisse der Messergebnisse visualisieren, damit Sie sie auf einen Blick sehen können. Daher extrahieren wir die Daten des Gesamtwerts jeder Gruppe, wenn die chemischen Substanzen grob klassifiziert sind. ..
Außerdem enthalten die diesmal verarbeiteten Daten eine Mischung aus zwei verschiedenen Einheiten. Da es sinnlos ist, verschiedene Einheiten zu vergleichen, werden wir diesmal die Daten für jede Einheit teilen. Im folgenden Code wird in der ersten Zeile jeweils die Einheit angegeben, und diejenigen, die in Lipid und Nass umgewandelt wurden, werden extrahiert, und dann werden nur die Variablen extrahiert, die mit Σ beginnen. In der zweiten Zeile werden Variablen, die mit Σ beginnen, aber nicht den Gesamtwert der Konzentrationen chemischer Substanzen darstellen, tatsächlich aus den Daten gelöscht.
data_lipid = df[(df["Unit"] == "ng/g lipid") & df["ChemicalName"].str.startswith("Σ")]
data_lipid = data_lipid[(data_lipid["ChemicalName"] != "ΣOH-penta-PCB") & (data_lipid["ChemicalName"] != "ΣOH-hexa-PCB")
& (data_lipid["ChemicalName"] != "ΣOH-hepta-PCB") & (data_lipid["ChemicalName"] != "ΣOH-octa-PCB")]
data_wet = df[(df["Unit"] == "ng/g wet") & df["ChemicalName"].str.startswith("Σ")]
data_wet = data_wet[(data_wet["ChemicalName"] != "ΣOH-penta-PCB") & (data_wet["ChemicalName"] != "ΣOH-hexa-PCB")
& (data_wet["ChemicalName"] != "ΣOH-hepta-PCB") & (data_wet["ChemicalName"] != "ΣOH-octa-PCB")]
Datenübersicht
Dies ist das Ergebnis der Verarbeitung mit Abschnitt 3-1 (nur für diejenigen, deren Einheit ng / g Lipid ist). Durch Löschen unnötiger Daten konnten nur 131 Daten für 6 chemische Substanzen extrahiert werden.
```python
data_lipid
```
|
MeasuredID |
SampleID |
ChemicalID |
ChemicalName |
ExperimentID |
MeasuredValue |
AlternativeData |
Unit |
Remarks_x |
ProjectID |
... |
| 4371 |
24782 |
SAA001903 |
CH0000033 |
ΣDDTs |
EXA000001 |
68000.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4372 |
24768 |
SAA001903 |
CH0000096 |
ΣPCBs |
EXA000001 |
5700.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4381 |
24754 |
SAA001903 |
CH0000138 |
ΣPBDEs |
EXA000001 |
170.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4385 |
24902 |
SAA001903 |
CH0000142 |
ΣHBCDs |
EXA000001 |
5.6 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4389 |
24810 |
SAA001903 |
CH0000146 |
ΣHCHs |
EXA000001 |
1100.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| ... |
... |
... |
... |
... |
... |
... |
... |
... |
... |
... |
... |
| 4812 |
25125 |
SAA001940 |
CH0000152 |
ΣCHLs |
EXA000001 |
160.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4816 |
25112 |
SAA001941 |
CH0000033 |
ΣDDTs |
EXA000001 |
9400.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4817 |
25098 |
SAA001941 |
CH0000096 |
ΣPCBs |
EXA000001 |
1100.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4818 |
25140 |
SAA001941 |
CH0000146 |
ΣHCHs |
EXA000001 |
41.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
| 4819 |
25126 |
SAA001941 |
CH0000152 |
ΣCHLs |
EXA000001 |
290.0 |
NaN |
ng/g lipid |
NaN |
PRA000030 |
... |
131 rows × 74 columns
Abschnitt 3-2 Löschen Sie nicht benötigte Spalten
Löschen Sie als Nächstes nicht benötigte Spalten. Wenn die Informationsmenge gleich ist, ist es im Allgemeinen besser, eine kleine Datenmenge zu verarbeiten, sodass unnötige Daten besser gelöscht werden. In dem diesmal behandelten Datenrahmen gibt es viele Leerzeichen ohne Daten. Dies wird in Python als N / A (Gleitkommazahl) behandelt, und obwohl keine Informationen vorhanden sind, nimmt es eine bestimmte Menge an Speicherplatz in Anspruch. Daher werden alle nicht zutreffenden Spalten (Spalten ohne Informationen) aus dem Datenrahmen gelöscht.
```python
data_lipid = data_lipid.dropna(how='all', axis=1)
data_wet = data_wet.dropna(how='all', axis=1)
```
Datenübersicht
Dies ist das Ergebnis der Verarbeitung von Abschnitt 3-2. Es ist ersichtlich, dass die Anzahl der Spalten (der Wert der Spalten, die am Ende der Tabelle angezeigt werden) erheblich kleiner ist als das Ergebnis von Abschnitt 3-1.
```python
data_lipid
```
|
MeasuredID |
SampleID |
ChemicalID |
ChemicalName |
ExperimentID |
MeasuredValue |
Unit |
ProjectID |
SampleType |
TaxonomyID |
... |
| 4371 |
24782 |
SAA001903 |
CH0000033 |
ΣDDTs |
EXA000001 |
68000.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4372 |
24768 |
SAA001903 |
CH0000096 |
ΣPCBs |
EXA000001 |
5700.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4381 |
24754 |
SAA001903 |
CH0000138 |
ΣPBDEs |
EXA000001 |
170.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4385 |
24902 |
SAA001903 |
CH0000142 |
ΣHBCDs |
EXA000001 |
5.6 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4389 |
24810 |
SAA001903 |
CH0000146 |
ΣHCHs |
EXA000001 |
1100.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| ... |
... |
... |
... |
... |
... |
... |
... |
... |
... |
... |
... |
| 4812 |
25125 |
SAA001940 |
CH0000152 |
ΣCHLs |
EXA000001 |
160.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4816 |
25112 |
SAA001941 |
CH0000033 |
ΣDDTs |
EXA000001 |
9400.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4817 |
25098 |
SAA001941 |
CH0000096 |
ΣPCBs |
EXA000001 |
1100.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4818 |
25140 |
SAA001941 |
CH0000146 |
ΣHCHs |
EXA000001 |
41.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4819 |
25126 |
SAA001941 |
CH0000152 |
ΣCHLs |
EXA000001 |
290.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
131 rows × 37 columns
Visualisierung von Kap.4 ΣDDTs
Durch die Verarbeitung in
Kapitel 3 haben wir Daten für jede der beiden Arten chemischer Substanzen vorbereitet. Hier werden wir zunächst visualisieren, wie die Daten über ng / g Lipid von ΣDDTs gestreut sind. </ p>
Abschnitt 4-1 Datenaufbereitung
Zur Visualisierung werden zunächst ΣDDTs-Daten aus data_lipid extrahiert.
```python
data_ddt = data_lipid[data_lipid["ChemicalName"] == "ΣDDTs"]
ddt_vals = data_ddt.loc[:, "MeasuredValue"]
data_ddt
```
|
MeasuredID |
SampleID |
ChemicalID |
ChemicalName |
ExperimentID |
MeasuredValue |
Unit |
ProjectID |
SampleType |
TaxonomyID |
... |
| 4371 |
24782 |
SAA001903 |
CH0000033 |
ΣDDTs |
EXA000001 |
68000.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4401 |
24783 |
SAA001904 |
CH0000033 |
ΣDDTs |
EXA000001 |
140000.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4431 |
24784 |
SAA001905 |
CH0000033 |
ΣDDTs |
EXA000001 |
140000.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4461 |
24785 |
SAA001906 |
CH0000033 |
ΣDDTs |
EXA000001 |
130000.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
| 4491 |
24786 |
SAA001907 |
CH0000033 |
ΣDDTs |
EXA000001 |
280000.0 |
ng/g lipid |
PRA000030 |
ST004 |
34892 |
... |
5 rows × 37 columns
Abschnitt 4-2 Grundlegende Statistiken
Da die Daten in Abschnitt 4-1 extrahiert wurden, werden die Basisstatistiken berechnet, um die Eigenschaften dieses Datensatzes zu erfassen. Grundlegende Statistiken sind einige Statistiken, die den Datensatz zusammenfassen. Dieses Mal werden die Gesamtzahl der Daten, der Durchschnitt, die Standardabweichung, der erste Quadrant, der Medianwert, der dritte Quadrant, der Minimalwert und der Maximalwert berechnet.
```python
count = ddt_vals.count()
mean = ddt_vals.mean()
std = ddt_vals.std()
q1 = ddt_vals.quantile(0.25)
med = ddt_vals.median()
q3 = ddt_vals.quantile(0.75)
min = ddt_vals.min()
max = ddt_vals.max()
count, mean, std, q1, med, q3, min, max
```
(24, 102137.5, 81917.43357743236, 41750.0, 70500.0, 140000.0, 9400.0, 280000.0)
In der oberen Zelle werden die Basisstatistiken einzeln berechnet, aber Pandas hat eine Funktion, die die Basisstatistiken zusammen ausgibt.
avgs = ddt_vals.describe()
avgs
count 24.000000
mean 102137.500000
std 81917.433577
min 9400.000000
25% 41750.000000
50% 70500.000000
75% 140000.000000
max 280000.000000
Name: MeasuredValue, dtype: float64
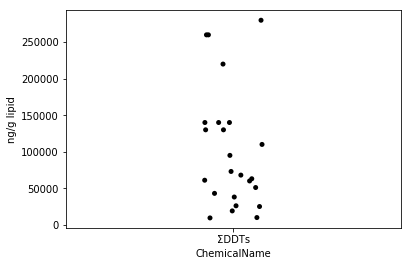
Sec.4-3 jitter plot
Zur Visualisierung der Streuung der Daten gibt es ein Streudiagramm, in dem alle Daten dargestellt werden (Eguchi: Ist dies ein Jitterdiagramm, kein Streudiagramm?). In Python können Sie mithilfe der Stripplot-Funktion von seaborn ganz einfach ein Jitter-Diagramm zeichnen. Das Jitter-Diagramm ist eine der guten Visualisierungsmethoden, mit denen jeder Himmel leicht beschrieben werden kann, indem den Punkten Schwankungen hinzugefügt werden, selbst wenn sich die Punkte überlappen.
fig = plt.figure()
ax = fig.add_subplot(1,1,1)
sns.stripplot(x="ChemicalName", y="MeasuredValue", data=data_ddt, color='black', ax=ax) #Zeichnung des Jitterplots
ax.set_ylabel("ng/g lipid")
plt.show()
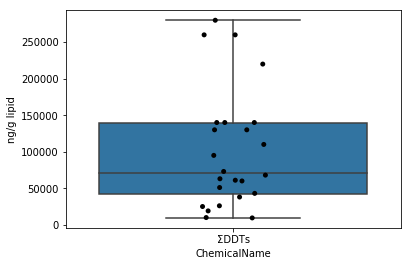
Abschnitt 4-4 Box Whisker
Im Gegensatz zu Abschnitt 4-3 zeigt das Box-Whisker-Diagramm eine Zusammenfassung des Datensatzes mit grundlegenden Statistiken. Die Box Whisker können mit der Boxplot-Funktion von Seaborn gezeichnet werden. Hier wird durch Überschreiben des Box-Whisker-Diagramms im vorherigen Jitter-Diagramm ein Diagramm ausgegeben, das das Verständnis der Verteilung der Daten erleichtert.
fig = plt.figure()
ax = fig.add_subplot(1,1,1)
sns.stripplot(x="ChemicalName", y="MeasuredValue", data=data_lipid[data_lipid["ChemicalName"] == "ΣDDTs"], color='black', ax=ax) #Zeichnung des Jitterplots
sns.boxplot(x="ChemicalName", y="MeasuredValue", data=data_lipid[data_lipid["ChemicalName"] == "ΣDDTs"], ax=ax) #Zeichnen einer Box Whisker
ax.set_ylabel("ng/g lipid")
plt.show()
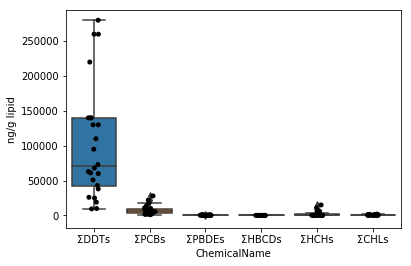
Visualisierung der Daten jeder chemischen Substanz von Kapitel 5 Snameri ( Neophocaena phocaenoides </ i>)
In Kapitel 4 wurde nur der Wert von ΣDDTs visualisiert. Wenden Sie es an, um data_lipid- und data_wet-Daten zu visualisieren.
### Abschnitt 5-1 Box Whisker (Anwendung von Abschnitt 4-4)
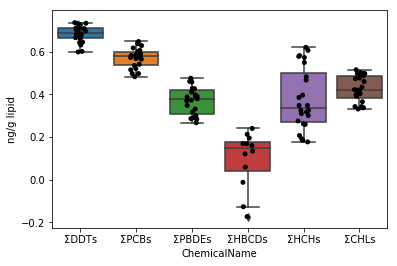
Zeichnen Sie zunächst ein Diagramm für die Daten von data_lipid auf dieselbe Weise wie in Abschnitt 4-4. Da die Stripplot / Boxplot-Funktion eine Funktion zum automatischen Klassifizieren von Daten anhand des dem Parameter x zugewiesenen Spaltennamens hat, reicht es aus, die gesammelten Daten den Parameterdaten unverändert zuzuweisen.
```python
fig = plt.figure()
ax = fig.add_subplot(1,1,1)
sns.stripplot(x="ChemicalName", y="MeasuredValue", data=data_lipid, color='black', ax=ax) #Zeichnung des Jitterplots
sns.boxplot(x="ChemicalName", y="MeasuredValue", data=data_lipid, ax=ax) #Zeichnen einer Box Whisker
ax.set_ylabel("ng/g lipid")
plt.show()
```
Abschnitt 5-2 Verbesserung von Abschnitt 5-1
In der obigen Grafik ist der Abstand der y-Achse groß, da die ΣDDTs stark variieren. Daher sind die verbleibenden 5 Diagrammtypen zerkleinert und schwer zu erkennen. Daher wird die Darstellung des Diagramms erleichtert, indem der logarithmische Wert der Daten verwendet wird.
Wenn Sie den Logarithmus verwenden, kann der Logarithmus von 0 nicht verwendet werden. Überprüfen Sie daher, ob der Datensatz 0 enthält. Bei Verwendung der in-Anweisung wird das Vorhandensein oder Fehlen des entsprechenden Werts in TRUE / FALSE zurückgegeben.
0 in data_lipid.loc[:, "MeasuredValue"].values
False
Da festgestellt wurde, dass der Datensatz in der oberen Zelle keine 0 enthält, kann der logarithmische Wert unverändert übernommen werden.
```python
data_lipid.loc[:, "MeasuredValue"] = data_lipid.loc[:, "MeasuredValue"].apply(np.log10)
```
C:\Users\masah\Anaconda3\lib\site-packages\pandas\core\indexing.py:543: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame.
Try using .loc[row_indexer,col_indexer] = value instead
See the caveats in the documentation: http://pandas.pydata.org/pandas-docs/stable/indexing.html#indexing-view-versus-copy
self.obj[item] = s
fig = plt.figure()
ax = fig.add_subplot(1,1,1)
sns.stripplot(x="ChemicalName", y="MeasuredValue", data=data_lipid, color='black', ax=ax) #Zeichnung des Jitterplots
sns.boxplot(x="ChemicalName", y="MeasuredValue", data=data_lipid, ax=ax) #Zeichnen einer Box Whisker
ax.set_ylabel("log(ng/g) lipid")
plt.show()
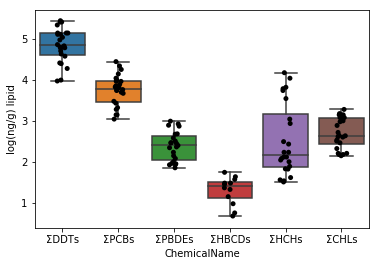
Visualisieren Sie
data_wet auf die gleiche Weise. Wenn der Datensatz 0 enthält, ersetzen Sie stattdessen 1/2 des Mindestwerts. Danach wird der Logarithmus aufgenommen und visualisiert. </ p>
if 0 in data_wet.loc[:, "MeasuredValue"].values:
data_wet["MeasuredValue"].replace(0, data_wet[data_wet["MeasuredValue"] != 0].loc[:, "MeasuredValue"].values.min() / 2) #Mindestwert 1/Ersatz 2
else:
pass
data_lipid.loc[:, "MeasuredValue"] = data_lipid["MeasuredValue"].apply(np.log10)
data_lipid.loc[:, "Unit"] = "log(ng/g lipid)"
fig = plt.figure()
ax = fig.add_subplot(1,1,1)
sns.stripplot(x="ChemicalName", y="MeasuredValue", data=data_lipid, color='black', ax=ax)
sns.boxplot(x="ChemicalName", y="MeasuredValue", data=data_lipid, ax=ax)
ax.set_ylabel("log(ng/g lipid)")
plt.show()
Fußnote
1 Es ist eine Funktion von Juyter Notebook, die als "magischer Befehl" bezeichnet wird, und es sollte beachtet werden, dass sie nicht im allgemeinen Python-Code verwendet werden kann.
2 Abkürzung für tabulatorgetrennte Werte. Es ist ein Dateiformat, in dem Daten durch TAB-Zeichen (am linken Ende der Tastatur) getrennt sind. Geeignet zum Speichern von tischförmigen Daten. Ein ähnliches Dateiformat ist CSV (Abkürzung für durch Kommas getrennte Werte, getrennt durch ", (Komma)").
