[PYTHON] Script to organize LDOS and PDOS from VASP output file DOSCAR
Intro
Good work. This is Hashimoto.
VASP can output LDOS and PDOS. LDOS (Local Density Of States) is the density of states of electrons belonging to a certain atom, and PDOS is the density of states of electrons belonging to each orbit. That is, the sum of LDOS is PDOS. The VASP result output file "DOSCAR" contains all-atom DOS and PDOS.
If you know the density of states, you can know which section of energy and how much the electrons occupy, so you can count the number of electrons that contribute to electrical conduction, physical quantities such as specific heat, and know the state of chemical bonds. I will. As you can see, it's a fairly important physical quantity, so I wrote my own Python script to organize the results while reading the file.
0. Premise
If you are not interested in manually organizing the contents of the results, for example, use pymatgen "https://pymatgen.org/" or p4vasp "http://www.p4vasp.at/#/". You can output DOS in a black box.
For pymatgen, the official reference and the article by RKS WEBSITE "http://ryokbys.web.nitech.ac.jp/pymatgen.html" will be helpful. The method for p4vasp is described on the VASP official website "https://www.vasp.at/wiki/index.php/CO_partial_DOS".
That said, honestly, it's subtle when asked if it's easy to use. In this article, the goal is "Because the format is not so difficult, extract DOSCAR with python and make it easy to understand."
1. Get DOSCAR
To get a DOSCAR that includes LDOS, you need to run the calculation twice (or three times).
Step1.1 First calculation
Prepare the files required for the calculation.
・ "POSCAR": After optimizing the structure, or creating the structure you want to calculate, prepare a file with a fixed structure.
・ "KPOINTS": Prepare a file with enough k points for the accuracy of the calculation to converge.
・ "POTCAR": Prepare a PAW (Projector Augmented Wave) method file to calculate all electrons.
・ "INCAR": Please refer to "https://qiita.com/youkihashimoto3110/items/de92172e0b5e9f3872d3" for the meaning of each tag used in the calculation setting file INCAR.
The first calculation is a new turn of the charge optimization (Electronic Relaxation) calculation. Comment out all the structural optimization parameters. Required tags are
ISTART = 0
PREC = Accurate
EDIFF = 1E-5
LREAL = Auto
ALGO = VeryFast
is. In the case of metal
ISMEAR = 1 #Or 2,-5
SIGMA = 0.5
In the case of insulators and semiconductors
ISMEAR = 0 #Or-5
SIGMA = 0.01
Use to adjust the SIGMA value so that the outcar entropy term is less than 1 meV per atom.
___ Do not use `ISMEAR> 0``` for insulators / semiconductors! ___ Also, using `ISMEAR = -5``` (tetrahedron method) improves accuracy, but is not suitable for most structures. (???)
Also, use "effective measures in case of non-convergence" as appropriate.
After completing the settings, execute the first calculation.
step2. Second calculation
When the calculation is completed, we will start the second calculation. In the second calculation, charge optimization is performed by the continuous job, so only the "INCAR" file is rewritten.
The changes from the first calculation file
ISTART = 1
ISYM = 0
LORBIT = 11
is. Let's recalculate the others in the same way.
Also, if you want to make the DOS energy step size finer,
EMIN = -10.0;EMAX = 17.0;NEDOS = 1001
Use the. The meaning of each is described in the template. `` `ICHARG = 11```, which provides a non-self-careless initial charge arrangement, is also useful.
2. Read DOSCAR
Open the "DOSCAR" output in the second calculation with any text editor.
The contents of DOSCAR are as shown in the figure below.
 I'm sorry it's hard to see. (The line breaks are due to ruby characters, but there are no line breaks in the real thing.)
VASP official "https://www.vasp.at/wiki/index.php/DOSCAR" and Tohoku University Professor Miyamoto "http://www.aki.che.tohoku.ac.jp/~taiko/vasp.html" Please refer to the website for information.
Also, the example shown above does not take spin into account in the calculation. In the case of calculation considering spins, it is also output for each spin.
I'm sorry it's hard to see. (The line breaks are due to ruby characters, but there are no line breaks in the real thing.)
VASP official "https://www.vasp.at/wiki/index.php/DOSCAR" and Tohoku University Professor Miyamoto "http://www.aki.che.tohoku.ac.jp/~taiko/vasp.html" Please refer to the website for information.
Also, the example shown above does not take spin into account in the calculation. In the case of calculation considering spins, it is also output for each spin.
3. Output the result with python
That is, "There are 6 fixed strings, first all DOS. There is the same string as the 5th line, LPDOS of the first atom has the same string as the 5th line, the second ...." You can see that it is the structure of. Also, there are no blank line breaks and you need to skip whitespace.
If you program based on these, it will be as follows.
Extract_dos.py
import csv
atom_index = [1,2,3] #The number of the atom for which you want to output LPDOS(Number from POSCAR)
doscar_name = "DOSCAR" #The name of the DOSCAR to read
save_csv_name = "doscar.csv" #Name to save DOSCAR as csv
save_dos = "dos.csv" #All DOS saved names
#Store in list from DOSCAR
with open(doscar_name, newline='') as doscar:
dos_read = csv.reader(doscar, delimiter=' ', skipinitialspace=True)
dos_list = [i for i in dos_read]
#Output DOSCAR as CSV
with open(save_csv_name, mode='w', newline='') as doscar_csv:
dos_write = csv.writer(doscar_csv)
dos_write.writerows(dos_list)
#Extract all dos from DOSCAR
i = 5 #Read start index
dos = []
count = 0
dos.append(dos_list[i])
while True:
i = i + 1
count = count + 1
dos.append(dos_list[i])
if dos[0] == dos_list[i+1]:
break
with open(save_dos, mode="w", newline='') as dos_csv:
dos_write = csv.writer(dos_csv)
dos_write.writerows(dos)
#Extract the specified lpdos from DOSCAR and save each in CSV
lpdos_all = []
while True:
i = i + 1
pdos = []
count = 0
pdos.append(dos_list[i])
while True:
i = i + 1
count = count + 1
pdos.append(dos_list[i])
if len(dos_list)==i+1:
break
if len(pdos[count]) != len(dos_list[i+1]):
lpdos_all.append(pdos)
break
if len(dos_list)==i+1:
break
#Save lpdos
for j in atom_index:
save_lpdos = "lpdos"+str(j)+".csv"
with open(save_lpdos, mode="w", newline='') as lpdos_csv:
lpdos_write = csv.writer(lpdos_csv)
lpdos_write.writerows(lpdos_all[j+1])
It's a terrible program, but I don't think it's hard to read.
atom_index=[]If you specify the number of the atom in the order of POSCAR, the LPDOS of that number will be displayed."lpdos_number.csv"It will be output as. After that, if you plot it with Excel etc., you can plot DOS in each atom and orbit.
In addition, the method of "Store in list from #DOSCAR" that removes spaces and stores in an array is effective when organizing VASP output files.
When using the above DOS, the result when ``` atom_index = [100,160,200]` `` is specified is shown.
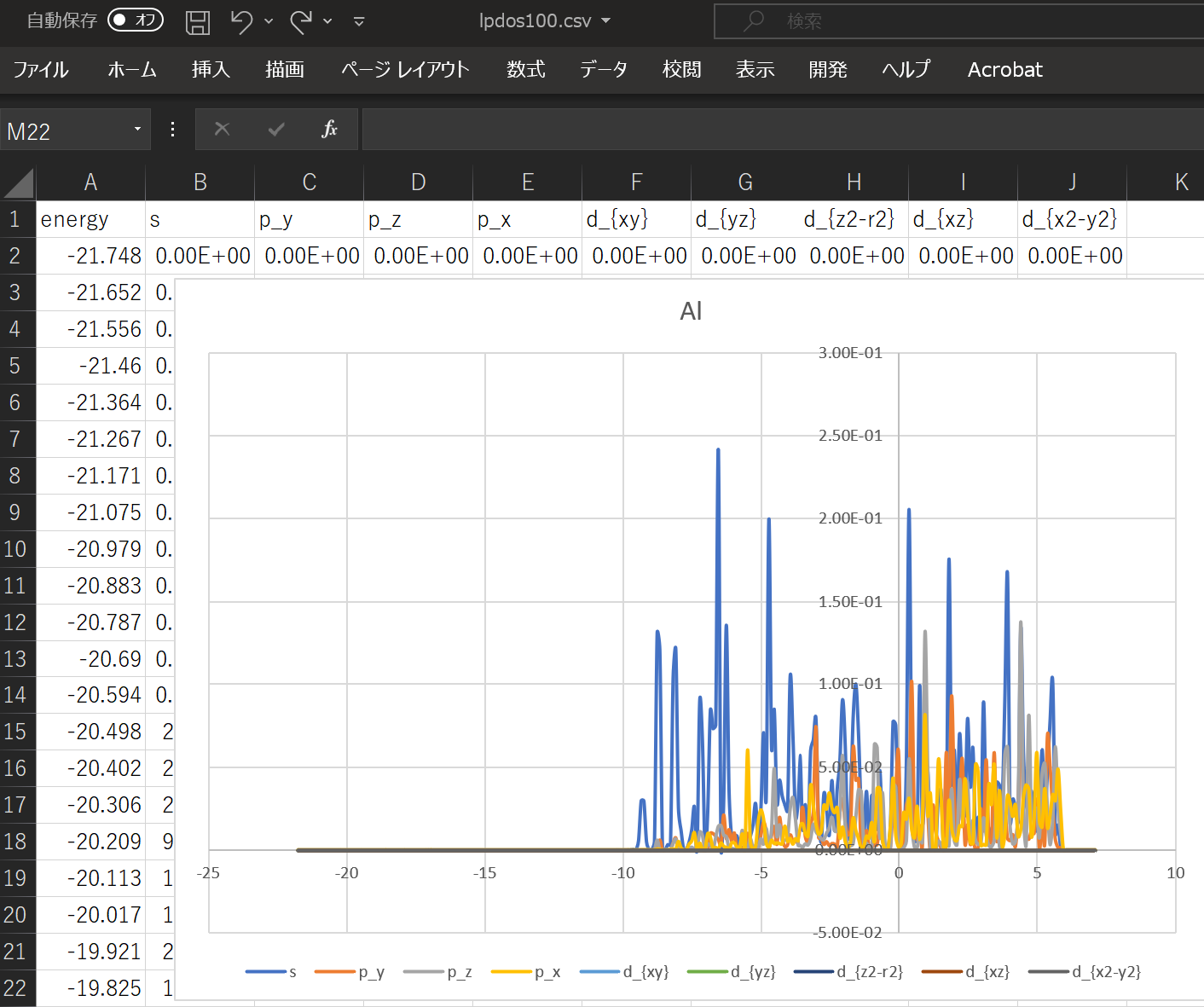
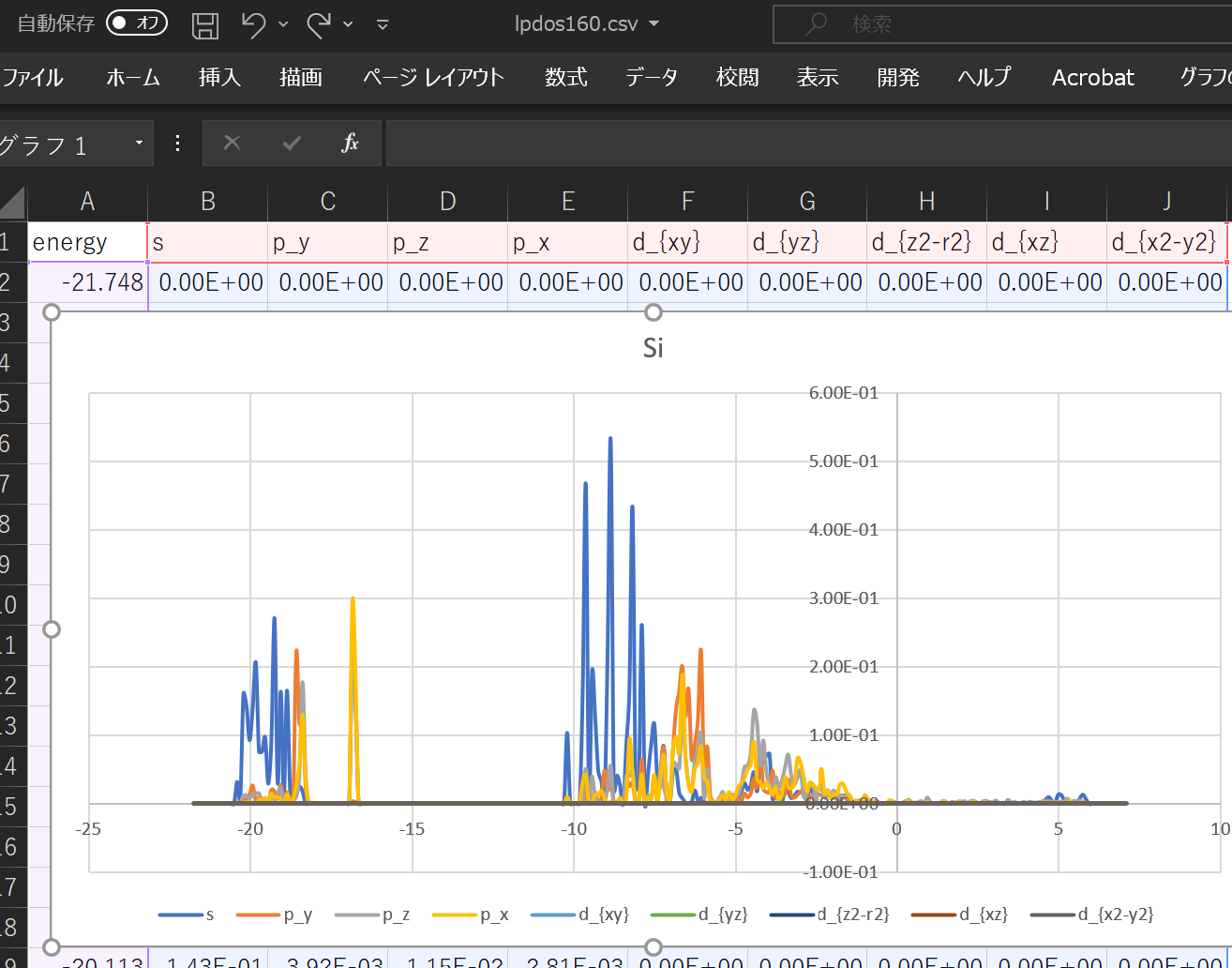
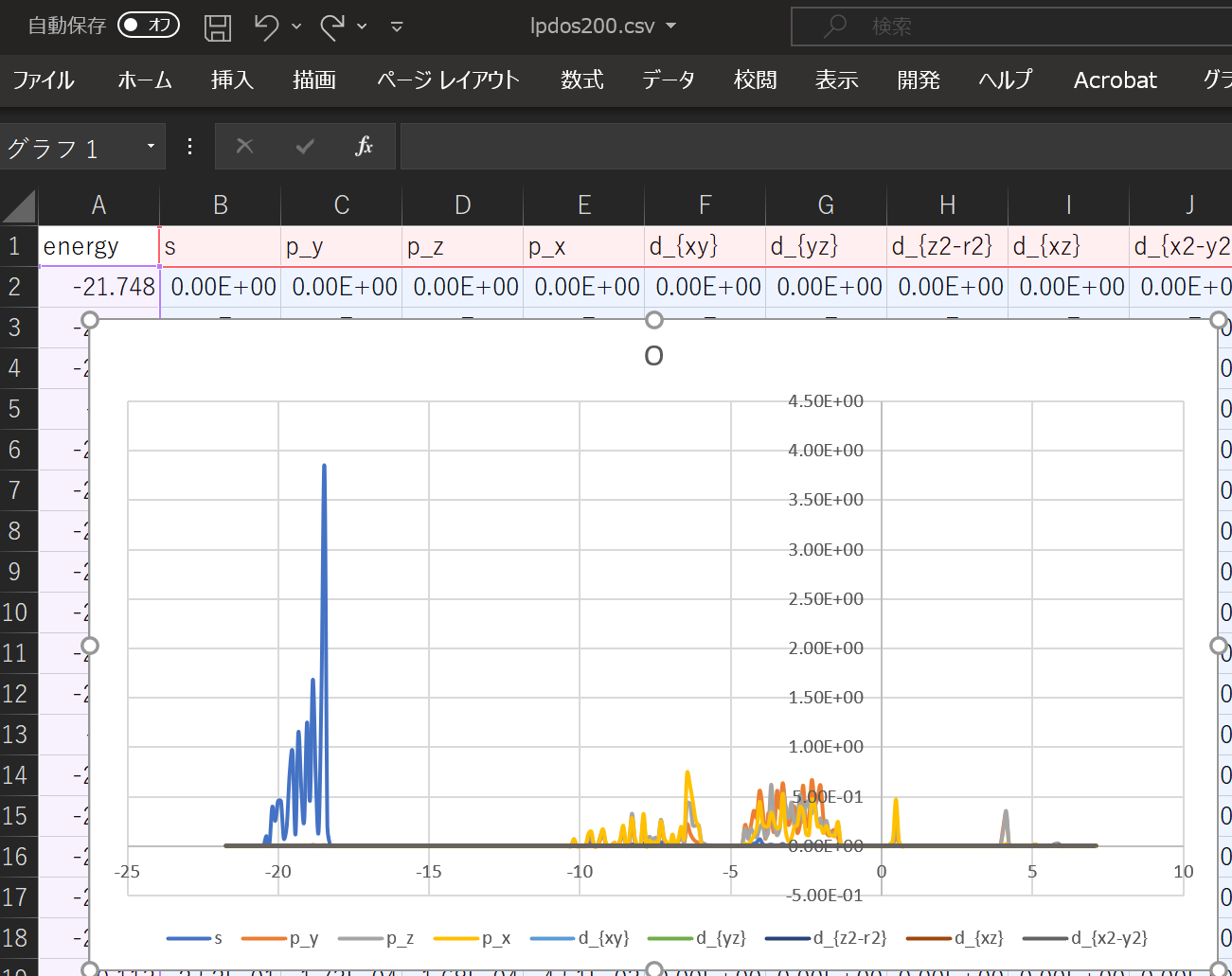
With this alone, you can predict what kind of structure and what kind of properties the substance has. ^^;
Recommended Posts